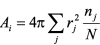 ,
,where rj: solvent accessible radius (van der Waals radius + 1.4) of atom j,
nj: number of points on atom j not occluded by other atoms,
N: number of distributed points on an atom.
Masato Masuya
Computing and Communications Center, Kagoshima University
masatom@biocomputing.cc
In computer simulation of proteins and nucleic acids, such as MD and/or MC, conformational energy + solvation energy calculation is needed. However, solvation energy calculation (implicit water, PB, RISM, etc.) is very time consuming. Therefore it can be applied to small system, or it can be included as implicit solvation effects, such as continuum model using distance dependent dielectric constant. While, there exist two methods that calculate solvation energy using solvent accessible surface area; ASP (atomic solvation parameters) model and GB/SA. In these methods, as solvent accessible surface area calculate fast, solvation energy could be calculate fast. So I decided to develop fast algorithm for molecular surface area calculation. NSOL is a fast solvation energy calculation program using fast molecular surface area calculation algorithm.
Solvent accessible surface is that part of the surface of a sphere centered at an atom with rvdW + rsol, where the center of a spherical solvent molecule can be placed in contact with the atomic van der Waals sphere (rvdW) without penetrating other atoms. Solvent accessible radius is defined as van der Waals radius + average radius of water molecule, usually 1.4 angstrom. Because of geometrical difficulty and finite accuracy of a computer, solvent accessible surface area has been calculated approximately. There are two method of calculation: an approximate analytical method and a finite accuracy numerical method.
| Atom type | Radius (Å = 10-10m = 10-1nm) | ||||
| (oons, jrf_) | (we92, sch3, sch4) | Bondi* | |||
| C | Aliphatic | 2.00 | 1.90 | 1.70 | |
| Aromatic | 1.75 | ||||
| Carbonyl/Carboxyl | 1.55 | ||||
| O | 1.40 | 1.40 | 1.52 | ||
| N | 1.55 | 1.70 | 1.55 | ||
| S | 2.00 | 1.80 | 1.80 | ||
| H | N/A | N/A | 1.20 | ||
| Water | 1.40 | 1.40 | N/A | ||
*Bondi, J.Phys.Chem., 68, 441, 1964.
Analytical:
Numerical:
Because of its stability, I adapt a numerical method.
Surface area of atom group i is obtained by,
where rj: solvent accessible
radius (van der Waals radius + 1.4) of atom j,
nj: number of points on atom j not occluded by other atoms,
N: number of distributed points on an atom.
It is permitted up to 12 points for distributing points equally. Several methods have proposed so far:
Saff's(The Mathematical Intelligencer, vol.19, p.5, 1997) distribution by energy minimization gives the best results.
To accelerate the computation, selected neighbors of a central atom to be removed from the computation preprocessing step. Such as;
NSOL 1.x adopt a method that uses artificial grid as the
same as Eisenharber et al. This is a standard approach in
computational geometry to group spatially close objects. In
difference of NSC, an implementation of DCLM, NSOL 1.x has an
optimized code for vector processor.
The DCLM method of Eisenharber et al. uses the second cube division for the points on sphere, but I used only spatial division.
NSOL 2.x adopt a method by sorting algorithm.
NSOL 3.x adopt my newest algorithm using mathematical morphology. In this method, distance calculation is not needed.
You can download each version of NSOL from following link:
| NSOL 1.7 | 2003/12/11 | Fortran 77, C* |
| NSOL 2.4 | 2003/12/22 | Fortran 77 |
| NSOL 3.0 |
* Compare to nsol.f, nsol.c is slightly slower and does not have vector optimization code.
Assuming that solvation energy is approximated in proportion to solvent accessible surface area.
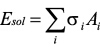
where σi: atomic solvation parameter,
Ai: solvent accessible surface area.
Several atomic solvation parameters have been proposed, such as, Ooi et al., Vila et al., Wesson et al., etc. And Temperature dependent solvation energy has been also proposed by Ooi et al.
oons: T. Ooi, M. Oobatake, G. Némethy, and H.A. Scheraga, Accessible surface areas as a measure of the thermodynamic parameters of hydration of peptides, Proc. Natl. Acad. Sci. USA (1987) vol.84, pp.3086-3090.
jrf_: J. Vila, R.L. Williams, M. Vásquez, and H.A. Scheraga, Empirical solvation models can be used to differentiate native from near-native conformations of bovine pancreatic trypsin inhibitor, PROTEINS: Structure, Function, and Genetics (1991) vol.10, pp.199-218.
we92, sch3: L. Wesson and D. Eisenberg, Atomic solvation parameters applied to molecular dynamics of proteins in solution, Protein Science (1992) vol.1, pp.227-235.
sch4: C.A. Schiffer, J.W. Caldwell, P.A. Kollman, and R.M. Stroud, Protein structure prediction with a combined solvation free energy-molecular mechanics force field, Molecular Simulation (1993) vol.10, pp.121-149.
Copyright © 1999-2010 Masato Masuya. All rights reserved.